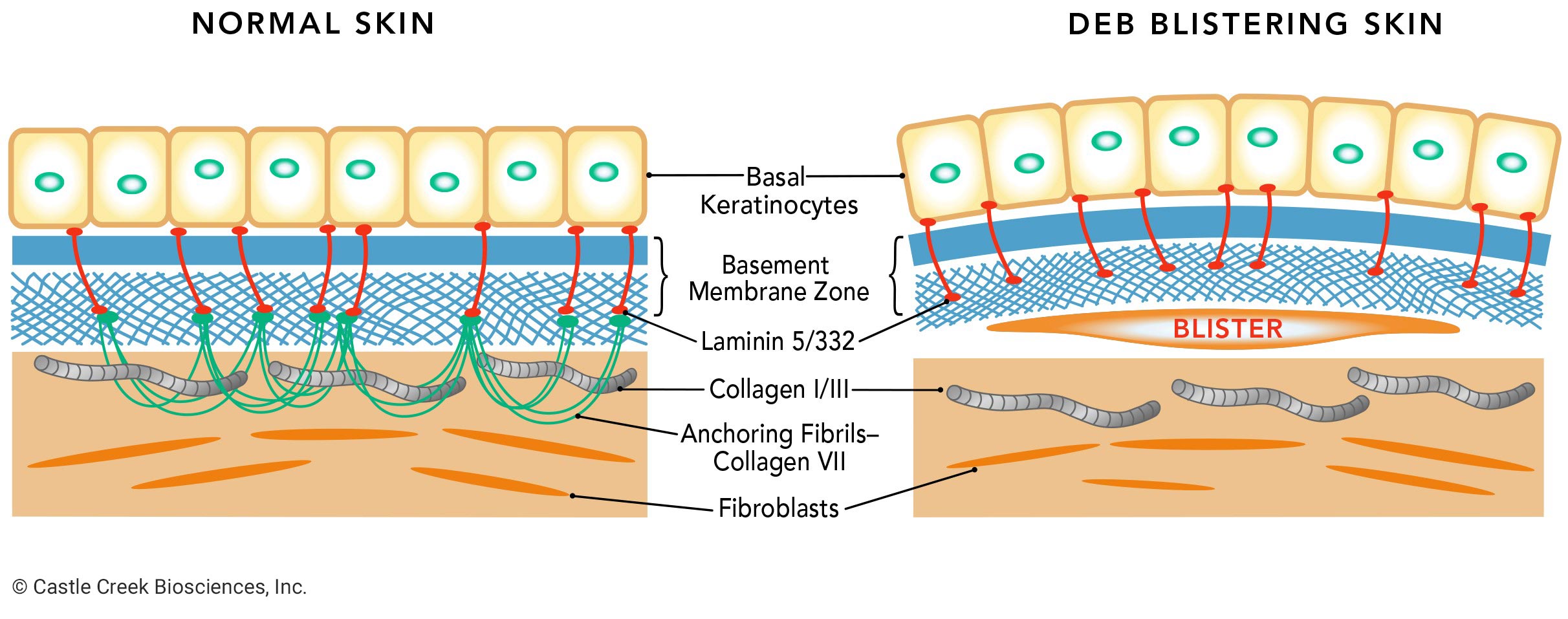
Dystrophic epidermolysis bullosa (DEB) is one of the four major types of epidermolysis bullosa (EB), a genetic connective tissue disorder that causes the skin to be extremely fragile. DEB results from mutations in the type VII collagen gene, or COL7A1, which encodes COL7. COL7—a protein—is the main component of anchoring fibrils, which anchor the basement membrane zone of the epidermis to the dermis, providing structural integrity to the skin at the dermal-epidermal junction.
When functional COL7 is deficient, the anchoring fibrils cannot form properly. Without these fibrils the skin layers separate, causing blisters and wounds in response to any kind of friction, like rubbing or scratching.
Recessive dystrophic epidermolysis bullosa (RDEB)—a severe form of DEB1—is a devastating progressive, painful and debilitating disorder. RDEB is an autosomal recessive disorder, meaning that a child has inherited copies of the defective gene from both parents3. Patients with RDEB typically show signs and symptoms shortly after birth and are diagnosed as newborns due to the severity of the disease. RDEB patients have the highest rate of morbidity and mortality of all genetic blistering disorders.
Dominant dystrophic epidermolysis bullosa (DDEB) patients typically have higher levels of COL7A1 expression, resulting in milder signs and symptoms with blistering often limited to the hands, feet, knees and elbows. Nevertheless, untreated symptoms may lead to serious complications, which could be fatal.
D-Fi, also known as FCX-007, (dabocemagene autoficel), is being developed as an autologous cell-based gene therapy to address the deficiency of functional COL7 in patients with dystrophic epidermolysis bullosa (DEB).
D-Fi has been clinically studied in a Phase 1/2 clinical study (NCT02810951), which assessed six patients with RDEB. In this study, 80% (8/10) of treated chronic wounds demonstrated at least 90% wound healing 12 weeks after the first injection of D-Fi. None of the untreated wounds were healed. D-Fi was well-tolerated post-administration with few reports of temporary redness or discoloration at the injection site.
The U. S. Food and Drug Administration has granted Orphan Drug designation to D-Fi for the treatment of Dystrophic Epidermolysis Bullosa, which includes RDEB. In addition, D-Fi has been granted Rare Pediatric Disease designation, Fast Track designation and Regenerative Medicine Advanced Therapy (RMAT) designation by the FDA for treatment of RDEB.
Dermal fibroblasts are collected from the patient, cultured and genetically modified to provide functional COL7 and then injected into the affected areas.
Explore Advocacy & Educational Resources
Castle Creek Biosciences’ product candidates are investigational. None of our investigational gene therapies have received marketing approval by the U.S. Food and Drug Administration or any other regulatory agency.
![]()